Devis Benfaremo, Cristina Dichiara, Gianluca Moroncini, Armando Gabrielli
Clinica Medica, Dipartimento di Scienze Cliniche e Molecolari, Università Politecnica delle Marche

Scenario Clinico
Giunge alla nostra osservazione una donna di 72 anni, che lamenta comparsa di fenomeno di Raynaud tetrapolare da circa 8-10 mesi. In anamnesi, la paziente riferisce pregressa abitudine tabagica, ipertensione arteriosa essenziale in trattamento, pregressi episodi di tachicardia sinusale inappropriata, pregresso pneumotorace spontaneo e crolli vertebrali multipli su base osteoporotica.
Quale approfondimento, il medico curante prescriveva l’esecuzione a domicilio dei seguenti accertamenti:
- Ricerca autoanticorpi, inclusi anticorpi anti-nucleo (ANA) ed anticorpi anti-antigeni nucleari estraibili (ENA): riscontro di ANA positività e anti-Scl70 positività ad alto titolo;
- Capillaroscopia periungueale: scleroderma pattern di tipo early. Assenti aree avascolari, densità capillare conservata, ectasia prevalentemente omogenea dei capillari (5-10%), alcuni aspetti di neoangiogenesi e rari depositi emosiderinici, flusso rallentato;
Alla luce della sintomatologia riferita, il medico specialista approfondisce l’anamnesi, dalla quale emergono nuovi elementi. La paziente riferisce infatti sensazione di gonfiore ed impaccio delle mani, al punto che ha dovuto rimuovere gli anelli. Inoltre, oltre al dolore secondario al fenomeno di Raynaud concomitano delle artralgie polidistrettuali migranti, in assenza di episodi sospetti per artrite.
Dopo aver raccolto l’anamnesi, il medico procede con l’esame obiettivo, che mostra edema bilaterale delle dita delle mani (puffy fingers) con ispessimento cutaneo (skin thickening) delle dita (Rodnan Skin Score modificato totale: 5). Non sono presenti ulcere acrali (digital ulcers) né esiti puntiformi a livello della punta delle dita (pitting scars). La paziente non presenta teleangectasie cutanee, mentre a livello polmonare è possibile obiettivare dei fini rantoli crepitanti bibasali.
Alla luce del quadro clinico, laboratoristico e strumentale, nonché dei reperti obiettivi, il medico definisce così il problema clinico: fenomeno di Raynaud tetrapolare associato a puffy fingers, artralgie diffuse, anomalie capillaroscopiche e ANA positività. Ritiene pertanto che l’ipotesi più probabile sia quella di una sclerosi sistemica (SSc, sclerodermia). Meno probabili, seppur da tenere in considerazione, sono la connettivite mista (MCTD), la connettivite indifferenziata o altre malattie specifiche del tessuto connettivo. Non merita di essere presa in considerazione, invece, l’ipotesi di un fenomeno di Raynaud primitivo.

Per tale motivo, si pone i seguenti quesiti diagnostici.
Quali sono gli aspetti clinico-anamnestici che orientano verso una forma secondaria di fenomeno di Raynaud? In una paziente con sospetta SSc, quali sono gli elementi che permettono di stabilire la diagnosi? Quali sono gli accertamenti imprescindibili nella valutazione iniziale di questi pazienti?
Il fenomeno di Raynaud è una condizione caratterizzata da vasospasmo delle piccole arterie e arteriole digitali scatenato dall’esposizione al freddo. Sebbene reversibile, la vasocostrizione è prolungata ed intensa, tale da provocare le tipiche modificazioni di colore delle dita durante l’attacco, dalla fase ischemica (bianco), alla fase cianotica (blu) fino alla fase iperemica (rosso).
Il fenomeno di Raynaud può essere primitivo o secondario (soprattutto a malattie autoimmuni sistemiche). Tale distinzione è molto importante in quanto la forma primitiva è generalmente benigna ed interessa soprattutto donne giovani con familiarità positiva per lo stesso fenomeno. Le forme secondarie, invece, vanno sospettate quando l’esordio è in età avanzata, nel sesso maschile, in caso di edema delle mani e/o ulcere cutanee e in presenza di alterazione degli esami di laboratorio o dell’esame capillaroscopico.
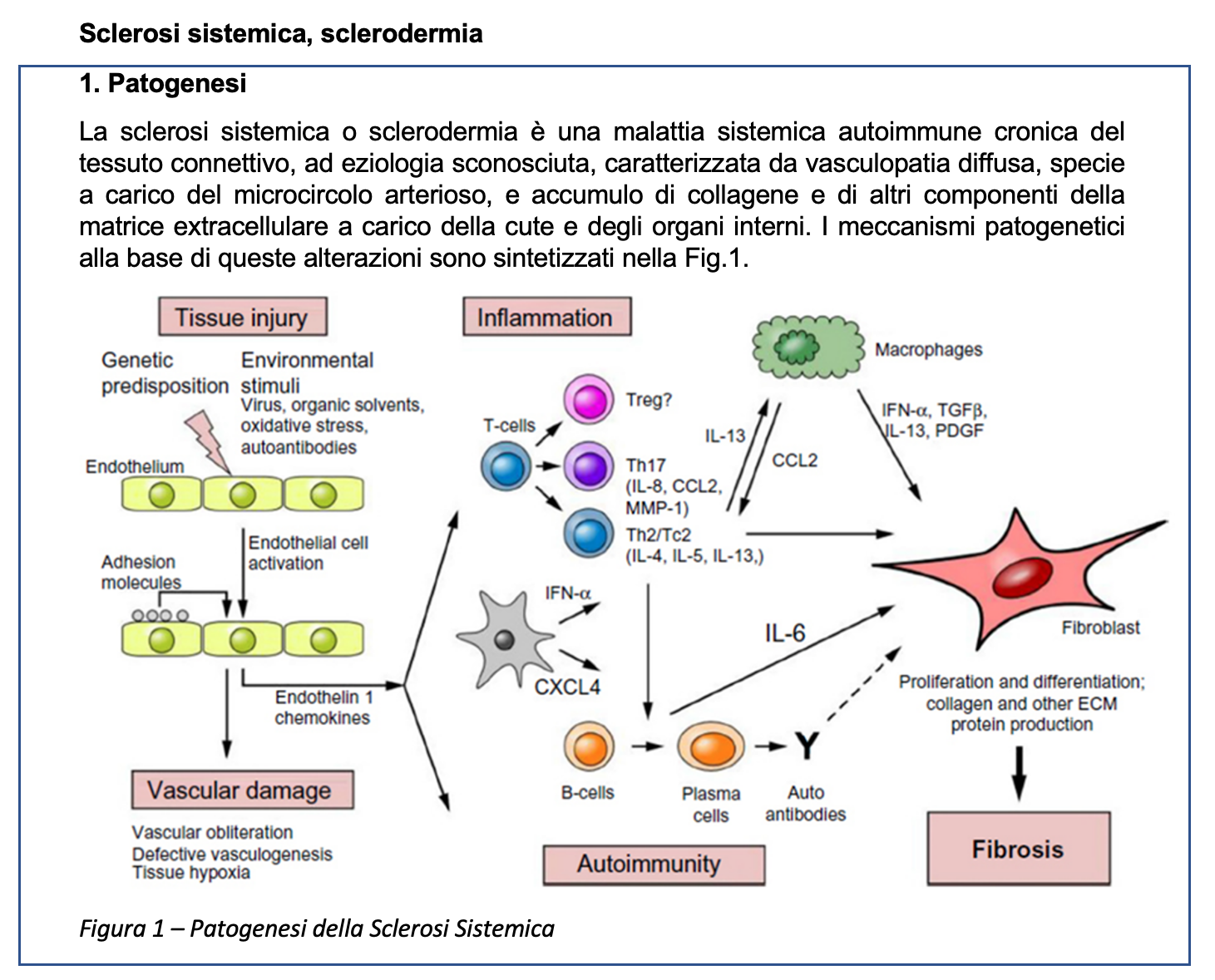
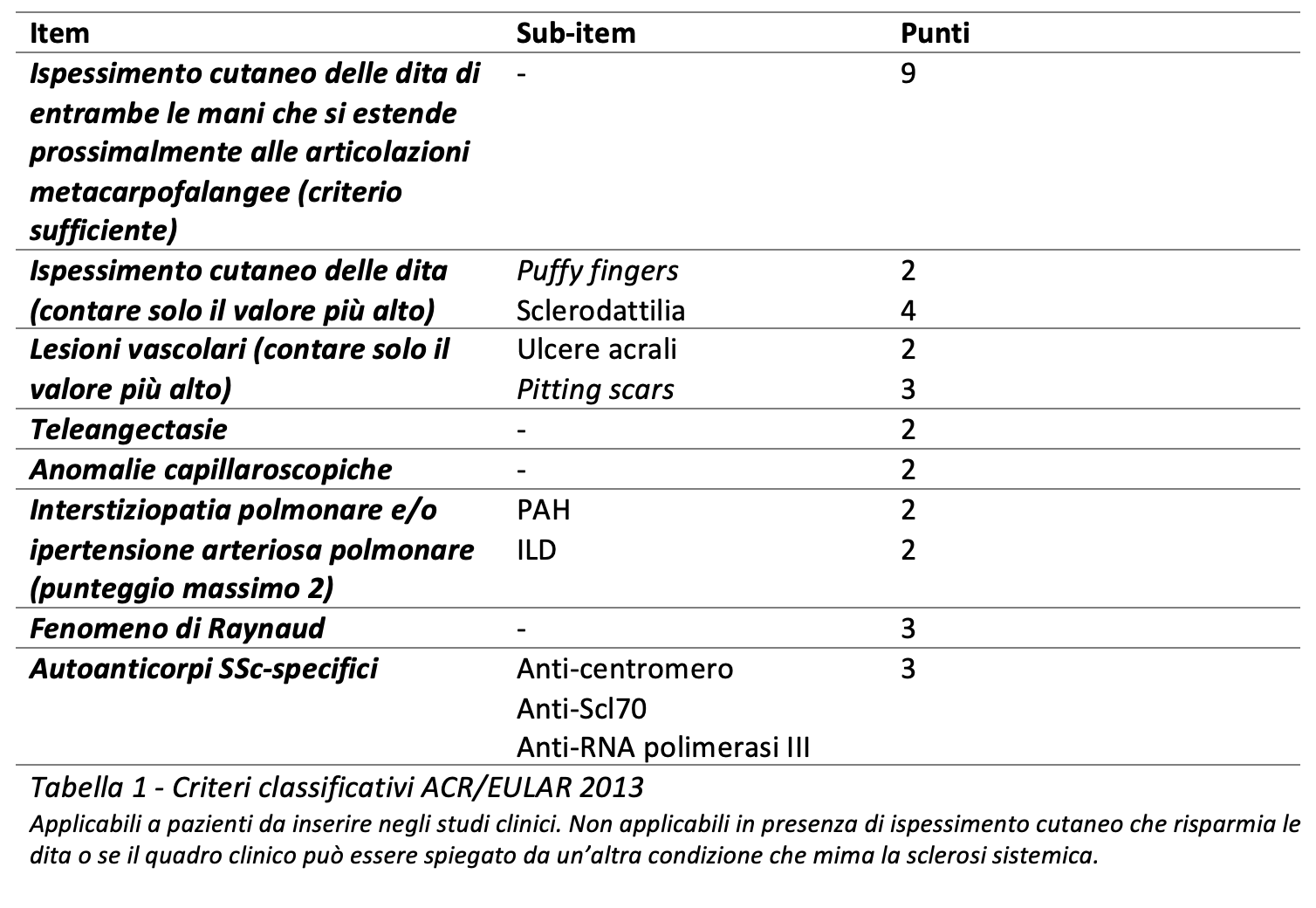
La diagnosi di sclerodermia si basa sul sospetto clinico, dopo aver valutato il quadro clinico-obiettivo, gli esami di laboratorio e quelli strumentali. Pur non essendo intesi come criteri diagnostici, bensì classificativi, i criteri dell’American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) del 2013 vengono spesso usati in ambiente reumatologico a supporto alla diagnosi clinica. La Tabella 1 riporta tali criteri classificativi. Possiamo quindi osservare che, pur non soddisfacendo il criterio sufficiente (ovvero la presenza di ispessimento cutaneo delle dita delle mani che si estende prossimalmente alle articolazioni metacarpo-falangee, criterio che da solo pesa 9 punti), la nostra paziente appare classificabile come affetta da SSc, alla luce della presenza di fenomeno di Raynaud, sclerodattilia, anomalie capillaroscopiche e presenza di anticorpi specifici, per un tale di 12 punti su un minimo di 9.
In aggiunta ad un anamnesi accurata, all’esame fisico, alla capillaroscopia periungueale e ai test di laboratorio, ulteriori indagini sono utili per confermare la diagnosi e determinare la presenza e l’entità del coinvolgimento sistemico. Dovrebbero far parte del work-up diagnostico iniziale:
- Test di funzionalità polmonare (PFT): identifica un difetto ventilatorio restrittivo o una diminuzione della capacità di diffusione del monossido di carbonio (DLCO).
- Imaging radiografico del polmone: la tomografia computerizzata ad alta risoluzione (HRCT) è preferita alla radiografia del torace per la migliore sensibilità. La HRCT rivela frequentemente anomalie polmonari interstiziali anche in pazienti con risultati PFT normali.
- Ecocardiografia Doppler: raccomandata per lo screening iniziale dell'ipertensione arteriosa polmonare (PAH).
Ulteriori indagini (ad es. manometria esofagea e/o anale, ricerca di colonizzazione batterica anomala dell’intestino) possono essere utili in base ai sintomi riferiti dal paziente.
Dopo circa 2-3 mesi dalla valutazione iniziale, la paziente viene pertanto ricoverata per eseguire gli approfondimenti del caso.
Dall’ultima valutazione la paziente riferisce comparsa di astenia progressiva e generalizzata, dispnea per sforzi moderati e lieve calo ponderale.
All’esame obiettivo si evidenzia la presenza di puffy fingers ed ispessimento cutaneo a pattern diffuso, con Rodnan Skin Score modificato totale pari a 27. L’obiettività toracica è caratterizzata dalla presenza di crepitazioni velcro-like bi-basali. Si riscontrano inoltre edemi declivi e una lesione ipercheratosica non ulcerativa peri-malleolare a destra.
Per indagare il coinvolgimento d’organo vengono richiesti i seguenti accertamenti:
- Spirometria con DLCO: insufficienza ventilatoria di tipo misto di grado moderato (FVC 81%) e riduzione della DLCO di grado moderato (60%).
- TC torace ad alta risoluzione senza e con mezzo di contrasto: modesto ispessimento interstiziale reticolare in sede basale bilateralmente, cui si associano areole di iperdensità parenchimale a vetro smerigliato, riferibili a segni di interstiziopatia fibrotica. Ipotonia esofagea.
- Ecocardiografia: disfunzione diastolica di primo grado, valvola aortica tricuspide sclerocalcifica, insufficienza mitralica e tricuspidalica di grado lieve da cui si stima una pressione arteriosa polmonare (PAPs) di circa 55 mmHg.
Alla luce di tali accertamenti, il medico di reparto si pone i seguenti quesiti.
In una paziente con diagnosi di SSc, quali sono i pazienti a più alto rischio di sviluppare una ipertensione arteriosa polmonare (PAH)? Quali sono le implicazioni cliniche e prognostiche di una PAH associata alla SSc? La stima ecocardiografica della pressione polmonare sistolica ha un valore sufficientemente accurato per porre diagnosi di ipertensione polmonare o è necessario procedere con ulteriori accertamenti?
L'ipertensione arteriosa polmonare (PAH) è una complicanza frequente della SSc, dal momento che coinvolge circa il 10-15% dei pazienti. La PAH è definita come una pressione media dell’arteria polmonare (mPAP) superiore a 25 mmHg a riposo (misurata mediante cateterismo cardiaco destro), con pressione di incuneamento capillare inferiore a 15 mmHg e resistenze vascolari periferiche superiori a 3 unità Wood.
I pazienti a rischio significativamente aumentato di PAH sono quelli con: variante cutanea limitata di lunga durata (lcSSC), ridotta DLCO (<80% del predetto) o un FVC/DLCO ratio superiore ad 1.6, negatività degli anticorpi anti-Scl70.
Da un punto di vista terapeutico, la terapia medica della PAH si avvale di tre categorie di farmaci: gli antagonisti dei recettori dell’endotelina, gli inibitori della 5-fosfodiesterasi e gli analoghi delle prostacicline.
La presenza di PAH è un fattore di rischio indipendente di mortalità nei pazienti con SSc.
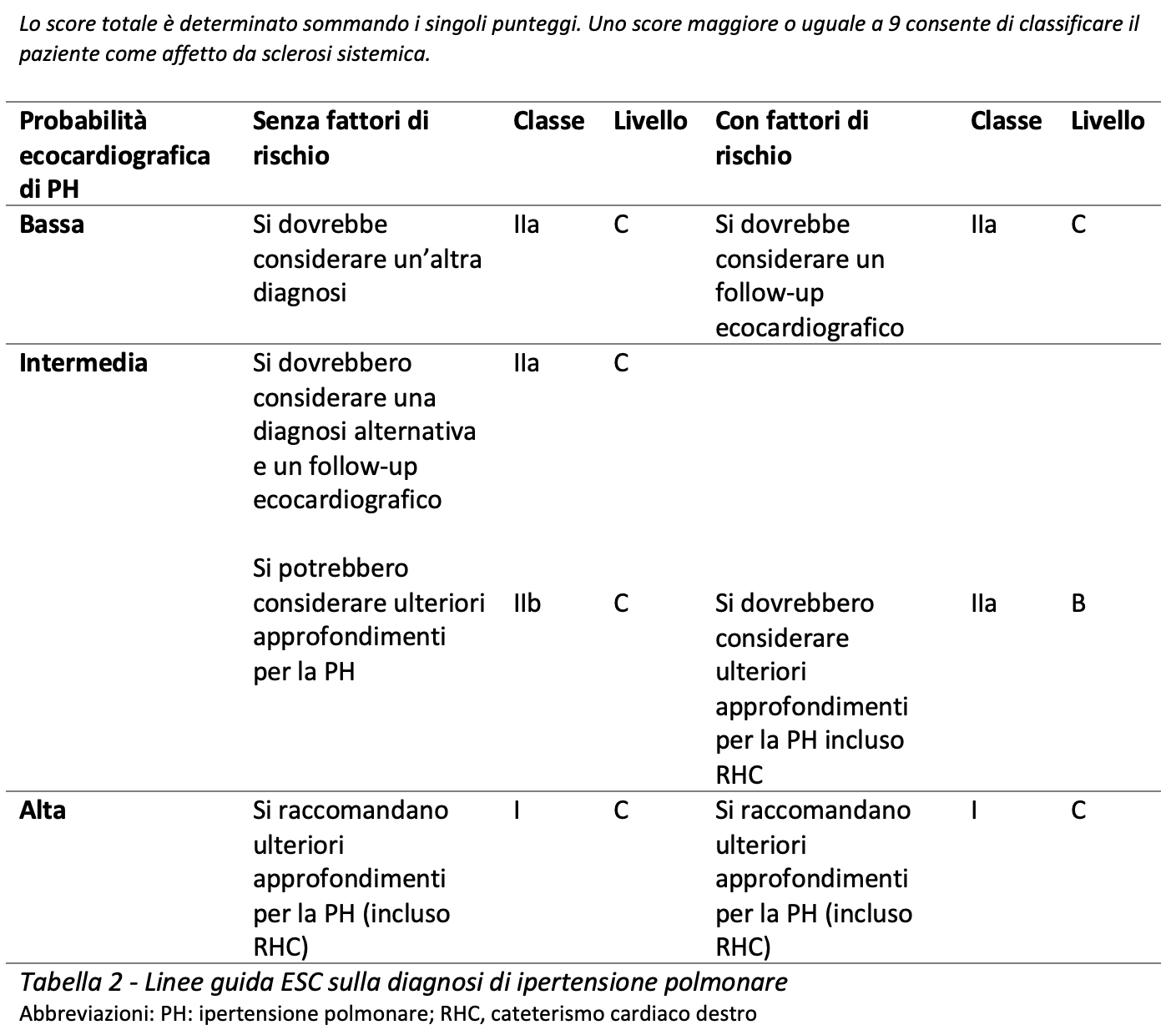
Per rispondere al terzo quesito, sono state prese in esame le linee guida della Società Europea di Cardiologia (2015) in merito all’Ipertensione Polmonare (Tabella 2). Secondo tali linee guida, quando interpretato nei diversi contesti clinici, l’esame ecocardiografico è indispensabile per determinare la probabilità di PAH e quindi la necessità di eseguire o meno il cateterismo cardiaco nei singoli pazienti. In un paziente con fattori di rischio per ipertensione polmonare, in presenza di una probabilità ecocardiografica intermedio-alta (definita in base ad una velocità di picco del rigurgito tricuspicalico superiore a 2.8 m/s), il cateterismo cardiaco destro deve essere preso in considerazione.
Per tale motivo, tenuto conto dei fattori di rischio della paziente, si decide di procedere con l’esecuzione in elezione del cateterismo cardiaco, che viene eseguito dopo circa un mese in prosecuzione di ricovero.
L’esame ha mostrato normali pressioni in atrio destro, ventricolo destro e arteria polmonare (pressione media 15 mmHg, pressione di incuneamento 12 mmHg). Normali le resistenze polmonari totali e arteriolari. Indice cardiaco a riposo conservato.
Sulla base dei dati raccolti, il sospetto di ipertensione polmonare non viene confermato e la diagnosi finale risulta essere: sclerosi sistemica variante diffusa ad interessamento cutaneo, polmonare ed esofageo.
Il medico si interroga pertanto in merito alla strategia terapeutica da adottare.
In una paziente con SSc, quali sono i fattori associati ad una prognosi peggiore? Quali sono le indicazioni al trattamento immunosoppressivo?
Nei pazienti con SSc rappresentano fattori prognostici sfavorevoli: il coinvolgimento cutaneo diffuso e progressivo, il coinvolgimento cardiaco e/o polmonare, la crisi renale sclerodermica, la presenza di anticorpi anti-Scl70, il sesso maschile e l’età più giovane all’esordio.
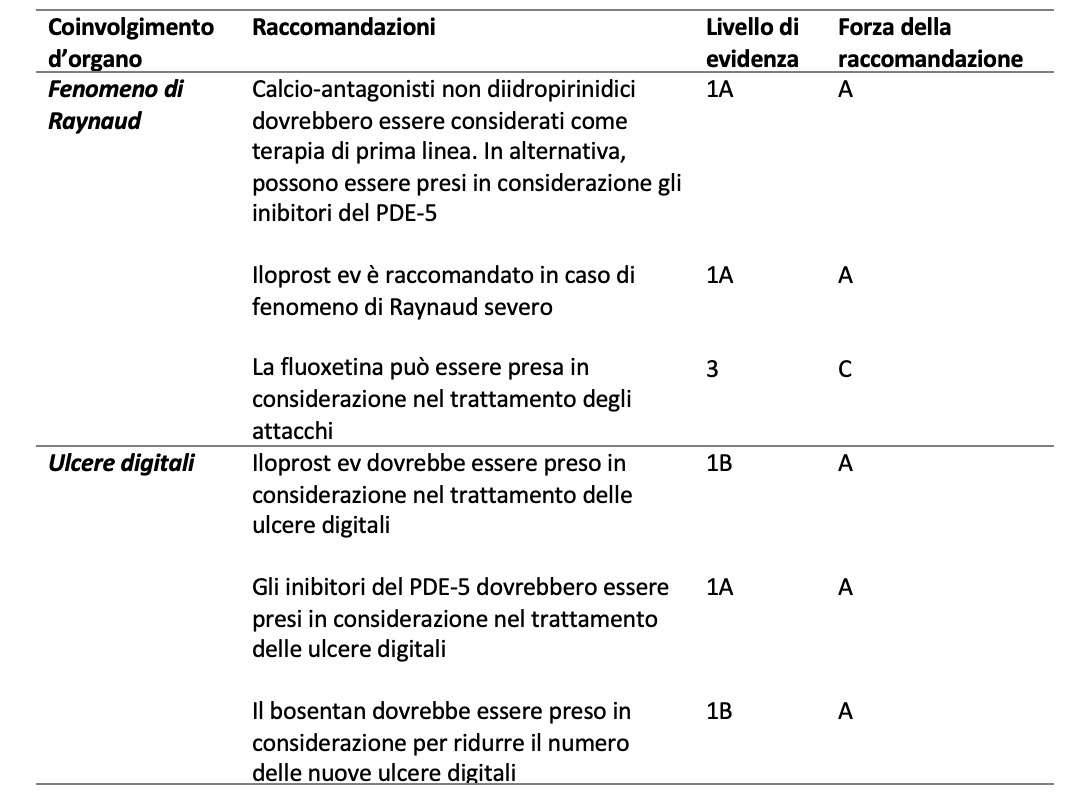
Per rispondere al quesito terapeutico sono state prese in esame le Raccomandazioni EULAR del 2017 (Tabella 3). Le linee guida sono state elaborate in modo da coprire i diversi domini coinvolti nella SSc: il fenomeno di Raynaud, le ulcere digitali, l’ipertensione arteriosa polmonare (PAH), la fibrosi cutanea e polmonare, la crisi renale sclerodermica ed il coinvolgimento gastrointestinale.
Per il trattamento della interstiziopatia in corso di SSc sono utilizzati farmaci immunosoppressori, in particolare la ciclofosfamide ed il micofenolato mofetile, quest’ultimo preferito in quanto gravato da minore tossicità sistemica. Dal 2019 è inoltre disponibile un nuovo inibitore delle tirosin chinasi, il nintedanib, che ha dimostrato superiorità rispetto al placebo nel rallentare il declino della funzionalità polmonare nei pazienti con SSc.
Nella nostra paziente, pertanto, in considerazione del coinvolgimento cutaneo progressivo (rapido peggioramento dello Score di Rodnan modificato) e della presenza di interstiziopatia polmonare, si decide di iniziare terapia immunosoppressiva con micofenolato mofetile 2 gr/die, in aggiunta alla bassa dose di steroide già in atto. Da un punto di vista gestionale viene programmata rivalutazione strumentale, a distanza di tre mesi, mediante esecuzione di spirometria e TC torace ad alta risoluzione.



La valigetta del medico
- La sclerosi sistemica (SSc) o sclerodermia è una malattia cronica rara a eziologia sconosciuta, di natura autoimmune, caratterizzata da anomalie vascolari microangiopatiche, alterazioni immunologiche e fibrosi diffusa. La SSc è più comune tra le donne rispetto agli uomini, specialmente nella fascia di età compresa tra 20 e 50 anni.
- I sintomi più frequenti comprendono fenomeno di Raynaud, poliartralgie, disfagia, dispnea, pirosi retrosternale, puffy fingers e fibrosi cutanea con sclerodattilia. Il coinvolgimento degli organi interni (specialmente esofago, tratto gastrointestinale inferiore, polmoni, cuore e reni) è una caratteristica che distingue la sclerosi sistemica dalle forme di sclerodermia localizzata ed è il principale responsabile della morbidità/mortalità.
- La diagnosi di sclerodermia si basa sul sospetto clinico, dopo aver valutato il quadro clinico-obiettivo, gli esami di laboratorio e quelli strumentali. I criteri dell’American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) vengono spesso usati a supporto alla diagnosi clinica.
- L'ipertensione arteriosa polmonare (PAH) è una complicanza frequente della SSc, dal momento che coinvolge circa il 10-15% dei pazienti. I pazienti a rischio significativamente aumentato di PAH sono quelli con: variante cutanea limitata di lunga durata, ridotta DLCO (<80% del predetto) o un FVC/DLCO ratio superiore ad 1.6, negatività degli anticorpi anti-Scl70.
- Nei pazienti con SSc rappresentano fattori prognostici sfavorevoli: il coinvolgimento cutaneo diffuso e progressivo, il coinvolgimento cardiaco e/o polmonare, la crisi renale sclerodermica, la presenza di anticorpi anti-Scl70, il sesso maschile e l’età più giovane all’esordio.
- Le Raccomandazioni EULAR sul trattamento della SSc sono state elaborate in modo da coprire i diversi domini coinvolti: il fenomeno di Raynaud, le ulcere digitali, l’ipertensione arteriosa polmonare, la fibrosi cutanea e polmonare, la crisi renale sclerodermica ed il coinvolgimento gastrointestinale.
BIBLIOGRAFIA
1. van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis. 2013 Nov;72(11):1747-55.
2. Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2016 Jan 1;37(1):67-119.
3. Kowal-Bielecka O, Fransen J, Avouac J, et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis. 2017 Aug;76(8):1327-1339.
4. Distler O, Highland KB, Gahlemann M, et al. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. N Engl J Med. 2019 Jun 27;380(26):2518-2528.
5. Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010 Oct;69(10):1809-15.