

Maria Giovanna Danieli, Veronica Pedini
Dipartimento di Scienze Cliniche e Molecolari, Sezione di Clinica medica
Facoltà di Medicina e Chirurgia, Università Politecnica delle Marche
La presentazione di un caso clinico esemplare fornisce l’occasione per ricordare gli elementi che fanno sospettare uno stato di immunodeficienza primitiva, in particolare la CVID e il modo ottimale per trattarla. Con particolare enfasi sull’uso corretto delle immunoglobuline sottocute.
Scenario clinico
Una donna di 40 anni inviata dal Pronto Soccorso giunge alla nostra osservazione per episodi recidivanti di otite catarrale presenti da circa quattro mesi e trattati con plurime linee di terapia antibiotica, con remissione dell’otalgia ma persistenza di febbricola serotina.
Dall’anamnesi emergono: piastrinopenia autoimmune trattata con corticosteroidi, immunoglobuline endovena e infine con splenectomia (2010); eradicazione di Helicobacter pylori; ipogammaglobulinemia di tutte le classi rilevata dal 2014; carcinoma papillifero della tiroide trattato con tiroidectomia e terapia radio-metabolica nel 2016; adenopatie mediastiniche e ascellari (referto istologico di due biopsie compatibile con quadro reattivo) e micronoduli polmonari nel follow-up; storia di dubbia allergia alla penicillina. All’esame fisico, modesto ingrandimento dei linfonodi superficiali a livello sottomandibolare bilateralmente ed ascellare a destra; non eritema né altri segni di flogosi all’ispezione del faringe. All’otoscopia membrana timpanica integra, assenza di essudato. Gli esami eseguiti nel Pronto Soccorso evidenziano leucocitosi neutrofila e alla radiografia del torace un addensamento para-ilare infero-posteriore destro in un quadro di diffusa accentuazione della trama interstiziale.
L’insieme degli elementi raccolti ci porta a definire così il problema:
Febbricola, infezioni ricorrenti a carico delle alte vie respiratorie e dell’orecchio medio e sospetta polmonite in paziente con storia di ipogammaglobulinemia, piastrinopenia autoimmune, pregresso carcinoma papillifero tiroideo e presenza di adenomegalie superficiali
Faceva spicco, tra i dati clinici e laboratoristici raccolti, il riscontro costante di un ridotto livello di immunoglobuline sieriche. Il primo compito è stato quindi quello di verificare la presenza o meno di un deficit anticorpale. L’esito dell’accertamento fu: IgG 247 mg/dl, IgA < 8 mg/dl, IgM 11 mg/dl.
Visto il contesto clinico - laboratoristico avanzammo due ipotesi diagnostiche:
- Infezioni ricorrenti a carico delle vie respiratorie in deficit anticorpale primitivo ad esordio nell’età adulta (immunodeficienza primitiva, PID; immunodeficienza comune variabile, CVID?)
- Infezioni ricorrenti a carico delle vie respiratorie in immunodeficienza secondaria (iatrogena? in corso di virosi sistemiche? da altre cause?)
Viene quindi programmato un piano di indagini comprendente lo studio fenotipico dei linfociti del sangue periferico per evidenziare un eventuale deficit maturativo dei linfociti B, il dosaggio degli anticorpi anti-tetano dopo vaccinazione, la sierologia per HIV, la ricerca di anticorpi anti CMV, la ricerca di CMV DNA e di EBV DNA, l’esame colturale dell’espettorato e, per le pregresse reazioni ai farmaci, una valutazione allergologica prima di impostare la terapia antibiotica.
Ci siamo posti alcuni quesiti.
Quali sono gli elementi che devono far porre il sospetto di un deficit anticorpale primitivo?
La Jeffrey Model Foundation (http://www.info4pi.org) un’associazione no profit dedicata a stimolare ricerche per migliorare la diagnosi e la cura delle PID, ha stilato i “campanelli d’allarme” per le immunodeficienze primitive nell’adulto, anche con l’intento di sensibilizzare la popolazione sull’opportunità di consultare il medico in presenza di una o più delle seguenti condizioni:
- Più di due episodi di otite in un anno
- Più di due episodi di sinusite in un anno, in un paziente non allergico
- Almeno una polmonite l'anno per più anni
- Diarrea cronica con calo ponderale
- Infezioni virali ricorrenti (raffreddore, herpes, verruche, condilomi)
- Frequente necessità di antibiotici per via endovenosa
- Ascessi ricorrenti della cute e degli organi interni
- Candidosi orale o cutanea persistente
- Infezioni da micobatteri atipici
- Storia familiare di immunodeficienza primitiva
Uno stato di immunodeficienza può essere inoltre sospettato ed in accordo con le indicazioni dell’ESID (European Society of Immunodeficiencies https://esid.org/), in presenza di alcune patologie non infettive come malattie autoimmuni (es. citopenie) o infiammatorie croniche (MICI), patologie linfoproliferative, sindrome orticaria/angioedema. Le manifestazioni di autoimmunità, di cui la più frequente è la piastrinopenia, possono interessare fino al 30% dei casi e rappresentano talora la modalità di esordio. Altre condizioni ricorrenti sono enteropatie, linfoproliferazioni, aumentato rischio di tumori, che sono appannaggio pressoché esclusivo dei deficit primitivi, mentre le citopenie immuni si possono riscontrare anche nelle immunodeficienze secondarie.
Gli esami eseguiti nella nostra paziente confermarono la marcata riduzione delle tre classi immunoglobuliniche; un deficit di maturazione dei linfociti B con assenza di cellule switched memory e accumulo di cellule naïve; debole produzione degli anticorpi anti-tetano dopo vaccinazione; assenza di alterazioni nel test di immunofissazione e nella ricerca di catene leggere nel siero e nelle urine.
Quali possono essere i fattori eventualmente responsabili di una immunodeficienza secondaria?
Di fronte ad un deficit anticorpale occorre sempre chiedersi se possa essere dovuto alla presenza di qualcuno dei fattori (Tab. 1) in grado di indurre una minor produzione di immunoglobuline.

Tabella 1 – Cause di ipogammaglobulinemia secondaria
Nella nostra paziente l’ipotesi di deficit secondario poteva essere ricondotta anche all’utilizzo cronico in passato di corticosteroidi per la piastrinopenia autoimmune, caso nel quale tuttavia il difetto interessa quasi esclusivamente la classe IgG e non si associa a deficit di maturazione dei linfociti B. La presenza di infezione da HIV, CMV, EBV non è stata confermata dagli esami virologici specifici; un linfoma è apparso altresì improbabile, considerata la storia di linfoadenomegalie da alcuni anni stabili nel tempo e la negatività dell’esame istologico; nessun elemento infine in favore di una malattia mielomatosa.
L’ipotesi di una forma secondaria non trovava pertanto riscontro negli accertamenti eseguiti.
Nell’ambito dei deficit primitivi quali sono i criteri diagnostici specifici per la CVID?
Nell’ambito dei deficit anticorpali primitivi la storia clinica di infezioni ricorrenti associate a citopenia autoimmune e sviluppo di neoplasia pone il sospetto di un deficit anticorpale del tipo immunodeficienza comune variabile, la più frequente immunodeficienza primitiva sintomatica dell’età adulta, con una prevalenza di 1/25.000-50.000 casi e maggior incidenza tra i 20 e i 40 anni. Nelle casistiche europee per questa affezione è segnalato un ritardo diagnostico importante, attorno agli otto anni.
Un ausilio alla diagnosi ci viene offerto dalla ricerca dei criteri diagnostici proposti dall’ESID del 1999, successivamente aggiornati nella revisione del luglio 2019 (Tab.2) (Seidel MG, et al. J Allergy Clin Immunol Pract 2019;7:1763).

Tabella 2 - Criteri ESID per la diagnosi di CVID (ESID Registry - Working definitions for clinical diagnosis of IEI, https://esid.org/)
La diagnosi di CVID poteva quindi considerarsi confermata.
Quesiti terapeutici
Quando va introdotta e con quali modalità una terapia sostitutiva con immunoglobuline?
Il cardine terapeutico dei deficit anticorpali è la terapia sostitutiva con immunoglobuline, ottenute dal plasma di migliaia di donatori contenente plurime specificità anticorpali che esprimono lo spettro di patogeni a cui questi ultimi sono stati esposti. I preparati di immunoglobuline umane normali attualmente disponibili in commercio sono costituiti quasi esclusivamente da anticorpi della classe IgG (circa il 96-98%). Per l’introduzione della terapia sostitutiva nei pazienti con ipogammaglobulinemia (Fig.1), di recente è stato proposto un algoritmo (Jolles S, et al. Clin Exp Immunol 2017;188:333).
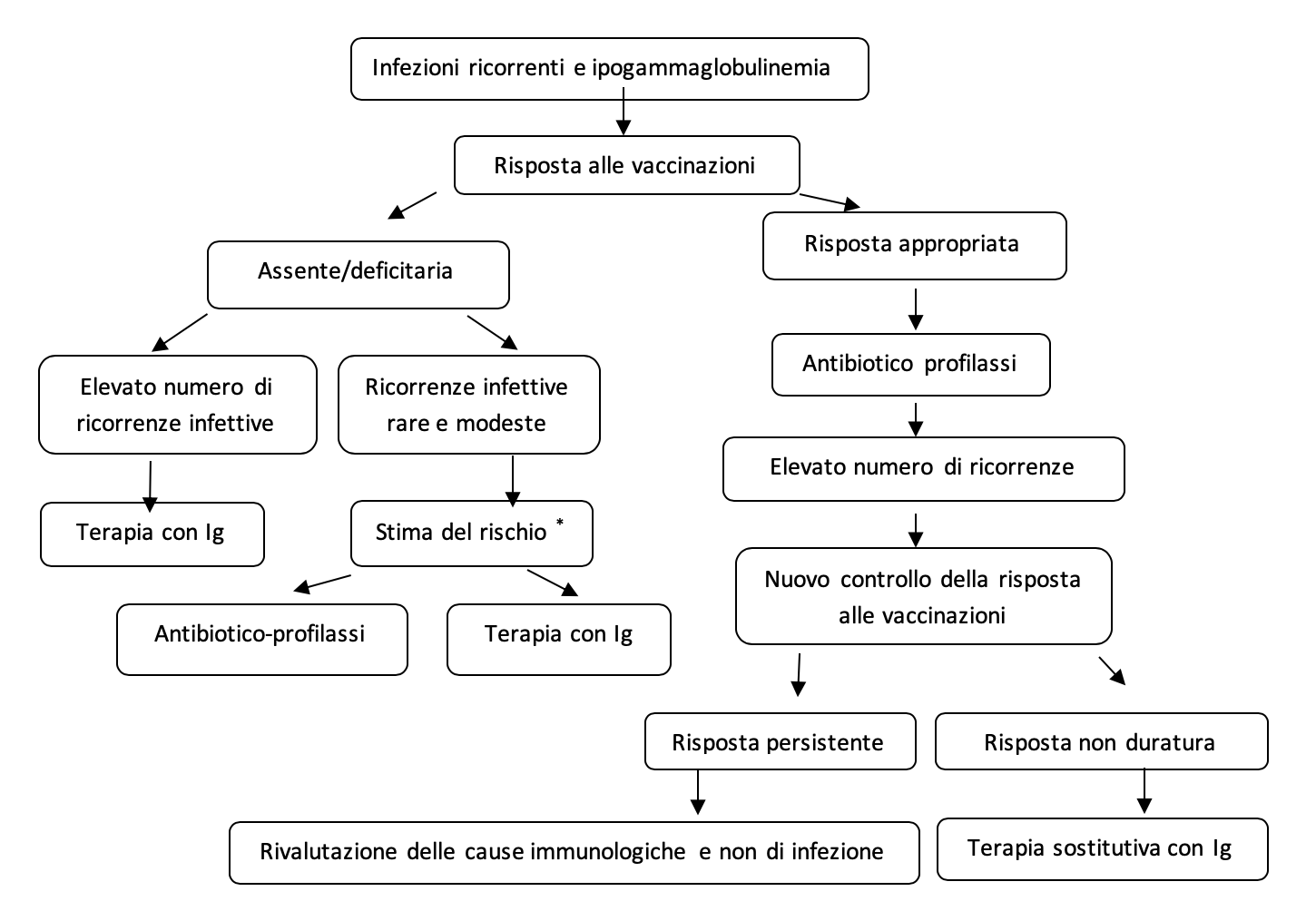
Figura 1 – Guida all’introduzione della terapia sostitutiva nei pazienti con ipogammaglobulinemia
*Valutazione del rischio globale di future infezioni nel singolo paziente, morbilità e mortalità sulla base di reperti clinici, laboratoristici e radiologici (evidenza di danno d’organo, di complicanze non infettive, alterata maturazione dei linfociti B, etc.). Nelle situazioni di non chiara definizione diagnostica, può essere utile un periodo di wait and see, con eventuale ciclo di antibiotico-profilassi e rivalutazione. Da Jolles S, et al. Clin Exp Immunol 2017;188:333, modificato.
Da questo algoritmo si evince come il livello di IgG non risulti più il criterio dirimente per decidere l’introduzione della terapia sostitutiva, ma assumano maggiore rilievo la mancata risposta vaccinale e l’entità delle ricorrenze infettive.
Il dosaggio standard delle Ig nella terapia sostitutiva è 400-600 mg/kg/mese. In genere dosaggi più bassi del range sono indicati nei soggetti senza complicanze, mentre dosaggi più elevati sono necessari in pazienti con complicanze della malattia quali bronchiectasie, malattia polmonare cronica ed enteropatia.
La terapia sostitutiva può essere somministrata per via endovenosa mediante infusioni mensili di immunoglobuline della durata di alcune ore da eseguire in strutture ospedaliere e presso day hospital dedicati, oppure per via sottocutanea mediante pompe infusive portatili da effettuare settimanalmente presso il proprio domicilio dopo adeguato addestramento ricevuto in ospedale. Di recente è stata poi introdotta la modalità sottocutanea facilitata che, associando alle immunoglobuline l’infusione di ialuronidasi, permette la somministrazione di maggiori volumi delle stesse per via sottocutanea, consentendo una cadenza mensile delle infusioni, eseguibili sempre a domicilio (Danieli M.G. et al. Immunotherapy 2016;8:995).
Considerata la cronicità della terapia sostitutiva (“terapia a vita”) con conseguente forte impatto sulla qualità di vita del paziente, la decisione del tipo di modalità di infusione va condivisa e spetta al singolo soggetto, salvo le eccezioni rappresentate da alcune controindicazioni mediche.
Il target da raggiungere non è più oggi rappresentato da un prefissato livello di IgG sieriche, l’obiettivo della terapia sostitutiva essendo quello di ottenere una significativa riduzione degli episodi infettivi annuali, fissato nelle diverse casistiche come inferiore a 2-3 episodi/anno.
Da ricordare infine che le immunoglobuline non hanno solo un ruolo meramente sostitutivo, ma possono espletare un’azione immunomodulante particolarmente utile nei pazienti con citopenie autoimmuni.
Quando è indicata una profilassi antibiotica?
Altra arma terapeutica nei deficit anticorpali è rappresentata dalla profilassi antibiotica che ha come scopo principale quello di ridurre la frequenza e la severità della ricorrenze infettive a carico delle vie respiratorie soprattutto nei pazienti con malattia polmonare cronica ed elevata frequenza infettiva. Tipici regimi di profilassi includono macrolidi (azitromicina) e possono essere somministrati durante tutto l’anno o nei periodi di incremento degli episodi infettivi, ad esempio quello invernale (Milito C, et al. J Allergy Clin Immunol 2019; 144:584).
Nella nostra paziente veniva eseguito ciclo di terapia antibiotica per la polmonite, con remissione della febbricola, ed iniziata terapia sostitutiva con immunoglobuline. Discusse le diverse opzioni di somministrazione con la paziente, si è optato per la terapia con immunoglobuline sottocute facilitate (20 g ogni quattro settimane), trattamento che ha comportato una significativa riduzione delle manifestazione infettive ricorrenti.