Maria Giovanna Danieli1,2, Cristina Mezzanotte3, Ilaria Claudi2
1 Clinica Medica
2 Scuola di Specializzazione in Allergologia ed Immunologia Clinica
3 Scuola di Specializzazione in Medicina Interna AOU Ospedali Riuniti, Università Politecnica delle Marche Ancona
Scenario clinico
Un giovane uomo di 27 anni giunge alla nostra osservazione per un quadro clinico caratterizzato da infezioni ricorrenti a carico di alte e basse vie aeree e delle vie genito-urinarie da circa 6 anni, con necessità ogni volta di far ricorso ad antibioticoterapia.
Dall’anamnesi emergono: nel 2012 linfoma non Hodgkin tipo Burkitt stadio IVB a localizzazione gastrica, midollare e testicolare, trattato con ciclofosfamide-vincristina-rituximab e adriblastina associati a citarabina e metotrexate intratecali e a radioterapia testicolare, seguiti da cicli di R-IVAC fino alla remissione; presenza di gozzo multinodulare e malattia da reflusso gastroesofageo.
Il paziente riconduce la comparsa delle ricorrenze infettive come successiva alla remissione completa della problematica neoplastica.
All’esame obiettivo epato-splenomegalia di I grado in assenza di linfoadenomegalie superficiali palpabili; al torace, riduzione del murmure vescicolare con fini crepitazioni teleinspiratorie alle basi polmonari; non altri reperti patologici di rilievo.
Negli esami ematici di routine non alterazioni significative, a parte una ipogammaglobulinemia segnalata all’elettroforesi siero-proteica; alle indagini endoscopiche e radiologiche nessuna apparente ripresa di malattia.
Il problema clinico per cui il paziente si è recato a visita può essere quindi ricondotto ad un quadro di infezioni ricorrenti a carico delle vie respiratorie e genito-urinarie, insorte da circa 6 anni a seguito di patologia linfomatosa chemio e radiotrattata. Dagli esami ematici il dato più rilevante è stato il riscontro di ipogammaglobulinemia, per cui si sono ulteriormente indagati i parametri immunologici del paziente.
Dall’immunodiffusione sono emersi i seguenti valori: IgG 347 mg/dl, IgA 53 mg/dl, IgM 70 mg/dl.
Sulla scorta del quadro clinico e della severa ipogammaglobulinemia, si sono formulate due principali ipotesi diagnostiche:
- Infezioni ricorrenti in deficit anticorpale primitivo ad esordio nell’età adulta (inborn errors of immunity (IEI); immunodeficienza comune variabile, CVID?);
- Infezioni ricorrenti in immunodeficienza secondaria (da malattia ematologica? iatrogena? altre cause, quali perdita renale o gastrointestinale?).
Le indagini immunologiche vengono a questo punto completate con la tipizzazione dei linfociti del sangue periferico, che mostra conta assoluta di linfociti B CD19+ entro il range di normalità, con “switched memory” ai limiti inferiori, in assenza di alterazioni della maturazione dei linfociti B.
Ulteriori approfondimenti richiesti sono stati il dosaggio degli anticorpi anti-tetano dopo vaccinazione risultati protettivi, oltre a sierologia per HIV, CMV DNA, EBV DNA, proteinuria della 24 ore, catene leggere libere sieriche ed urinarie, proteinuria di Bence-Jones, calprotectina fecale, risultati tutti non patologici.
Alla luce delle indagini effettuate, il quadro è apparso compatibile con un deficit immunitario di natura secondaria, che ha generato i seguenti quesiti:
Quesiti diagnostici
1. Quali sono le principali cause e fattori di rischio di deficit anticorpale secondario?
Le cause e i fattori di rischio di immunodeficienza secondaria possono essere molteplici (vedi Tab.1 e Tab.2). Le più comuni sono quelle legate a disordini ematologici quali la leucemia linfatica cronica, il mieloma multiplo e i linfomi. Il deficit anticorpale di natura iatrogena è anch’esso frequente perché si può sviluppare in seguito a trattamenti farmacologici diretti contro i linfociti B o con immunosoppressori quali rituximab, ciclofosfamide, corticosteroidi ad alte dosi, metotrexate, ecc… In altri casi invece alla base dell’immunodeficit vi è un’aumentata perdita delle immunoglobuline a livello renale, gastrointestinale o cutaneo. Cause più rare possono essere la malnutrizione, il trapianto di organi solidi e l’infezione da HIV.
Nel caso del nostro paziente abbiamo considerato il deficit anticorpale come secondario al disordine ematologico sviluppato in giovane età e ai trattamenti immunosoppressivi necessari per l’ottenimento della remissione completa da malattia (rituximab, ciclofosfamide, metotrexate, …) (Patel SY, 2019).
|
Tabella 1. Principali cause di deficit anticorpale secondario |
|
Disordini ematologici |
|
Leucemia linfatica cronica, linfomi, mieloma multiplo, leucemia mieloide acuta o cronica, sindromi mielodisplastiche, macroglobulinemia di Waldenstrom, amiloidosi |
|
Iatrogena |
|
Terapie contro i linfociti B (anti-CD20, rituximab, veltuzumab; anti-CD22, epratuzumab; anti-CD19, blinatumomab, CAR-T cells; anti-CD52, alemtuzumab; anti-CD38, daratumumab; anti-BAFF, belimumab), corticosteroidi, bortezomib, micofenolato mofetile, metotrexate, agenti alchilanti, sulfasalazina, sali d’oro, inibitori della tirosin chinasi, analoghi delle purine, anti-epilettici |
|
Trapianto |
|
Trapianto di cuore, rene, polmone, fegato, cellule staminali ematopoietiche |
|
Perdita di proteine |
|
Perdita renale (sindrome nefrosica), perdita gastrointestinale (malattie infiammatorie croniche intestinali, linfangiectasia intestinale, celiachia, infezioni intestinali, pericardite costrittiva, sindromi genetiche rare), perdita cutanea (ustioni o dermatiti severe) |
|
Circolazione linfatica |
|
Linfangiectasia intestinale, sindrome di Proteus, sindrome di Noonan, chilotorace |
|
Altro |
|
Plasmaferesi, distrofia miotonica, malnutrizione, HIV, dialisi peritoneale |
|
Tabella 2. Fattori di rischio per ipogammaglobulinemia iatrogena |
|
2. Quali possono essere i sintomi sviluppati dal paziente con immunodeficienza secondaria?
Lo spettro delle manifestazioni cliniche può essere molto vario: alcuni pazienti risultano del tutto asintomatici, nonostante valori di Ig estremamente bassi, altri manifestano una suscettibilità alle infezioni modesta, alcuni sviluppano invece complicanze infettive di severa entità. Tra le principali ricorrenze infettive vi sono quelle a carico delle alte e basse vie aeree e del tratto genito-urinario, sinusiti, sepsi, meningiti o infezioni da microrganismi opportunisti.
I principali batteri responsabili sono Haemophilus influenzae, Staphylococcus aureus, Streptococcus pneumoniae, Klebsiella pneumoniae; anche i funghi come la Candida sono possibili agenti eziologici.
Le citopenie immuni possono rappresentare un’altra manifestazione clinica nei deficit anticorpali secondari, in particolar modo l’anemia emolitica o la piastrinopenia autoimmuni (Patel SY, 2019).
3. Quali sono i principali step nel percorso diagnostico e assistenziale?
Un periodico follow-up dei pazienti esposti ai fattori di rischio menzionati sopra è essenziale per una diagnosi precoce di immunodeficienza e delle possibili complicanze infettive (Fig. 1). In particolar modo, nei pazienti ad alto rischio è indicato monitorare annualmente i valori di Ig sieriche e testare la risposta alle vaccinazioni in caso di bassi livelli anticorpali iniziali. Lo studio fenotipico dei linfociti da sangue periferico può essere utile per la diagnosi differenziale con forme di immunodeficienza primitiva, quale la CVID, in cui sono presenti alterazioni nella maturazione delle cellule B con conseguente accumulo di linfociti B “naive” e “unswitched”.
Vi sono poi procedure di screening particolari sulla base della natura dell’immunodeficienza. Nei pazienti trattati con idelalisib vanno monitorati segni e sintomi di infezioni respiratorie, ricercata una eventuale infezione da CMV e prescritta terapia profilattica contro l’infezione da Pneumocystis jirovecii.
Nei pazienti sottoposti a trapianto, l’ipogammaglobulinemia (IgG <6 g/L) o il consumo del complemento sono considerati fattori di rischio indipendenti per infezioni batteriche e da CMV, soprattutto se presenti contemporaneamente. Bassi titoli anticorpali contro il CMV e contro il polisaccaride pneumococcico riscontrati dopo il trapianto sono indicativi di una alterata risposta anticorpale specifica, per cui questi pazienti potrebbero beneficiare di una terapia sostitutiva con Ig. Oggigiorno è anche possibile analizzare la risposta CD8 CMV-specifica per valutare il rischio di infezione.
Nei pazienti sottoposti a trapianto la vaccinazione non è una pratica comune nei primi mesi dopo l’intervento (quando il rischio infettivo è severo), poiché la risposta anticorpale specifica dopo l’immunizzazione è bassa in questo periodo; inoltre i titoli anticorpali contro lo pneumococco e il CMV tendono gradualmente a ridursi durante il primo anno dal trapianto a causa della terapia immunosoppressiva, incrementando così il rischio infettivo (Patel SY, 2019).
Quesiti terapeutici
1. Quali strategie terapeutiche e di profilassi utilizzare nelle immunodeficienze secondarie?
Le linee guida raccomandano la profilassi antibiotica come terapia di prima linea per i pazienti con immunodeficienze secondarie suscettibili di infezioni gravi e nei pazienti sottoposti a chemioterapia o altri trattamenti immunosoppressivi. In genere lo schema suggerito si basa sulla somministrazione di azitromicina, 250 mg per os per tre giorni alla settimana. Se non si ottiene una risposta ottimale con la somministrazione di più linee di antibioticoterapia per via orale, si può procedere al trattamento antibiotico per via endovenosa (IVAB). Gli antibiotici nebulizzati e l'IVAB intermittente sono opzioni utilizzate principalmente nei pazienti affetti da bronchiectasie o con colonizzazione da Pseudomonas.
Secondo le linee guida della European Society for Medical Oncology (ESMO) del 2015 la profilassi antibiotica e antivirale va intrapresa nei pazienti con infezioni ricorrenti e/o a rischio infettivo molto elevato (ad es. profilassi contro lo Pneumocystis con cotrimossazolo durante il trattamento con chemio-immunoterapia).
Per quanto riguarda i pazienti trapiantati d’organo, le linee guida internazionali sulla infezione da CMV, pubblicate dal Transplantation Society International CMV Consensus Group, raccomandano l’utilizzo di terapia sostitutiva con immunoglobuline CMV-specifiche e raccomandano la profilassi antivirale per i pazienti ad alto rischio come i riceventi di trapianto di polmone (Cinetto F, 2021).
Un’altra strategia nei pazienti con deficit immunitario secondario è rappresentata dalle vaccinazioni profilattiche con virus inattivati.
Gli studi suggeriscono che la vaccinazione in una fase precoce (cioè prima dell'inizio della chemioterapia e della comparsa di ipogammaglobulinemia) può essere maggiormente utile nella generazione della memoria immunologica quando i livelli di anticorpi specifici sono bassi (Dhalla F, 2015). Inoltre, la valutazione dei tassi anticorpali specifici post-vaccinazione è funzionale sia dal punto di vista terapeutico (se vengono raggiunti livelli protettivi), sia nella stratificazione del rischio prima e dopo il trattamento. Sebbene sia generalmente approvato che i vaccini inattivati possono essere somministrati a pazienti con deficit anticorpale secondario, molte vaccinazioni non sono richieste per i pazienti che sono trattati con Ig, poiché livelli protettivi di anticorpi sono già presenti nei preparati di immunoglobuline.La vaccinazione antinfluenzale rappresenta un'eccezione, in quanto questi vaccini sono riformulati annualmente, quindi la protezione fornita dalla terapia sostitutiva è meno probabile.
Tuttavia, anche se la risposta anticorpale dopo la vaccinazione non è ottimale, i pazienti con immunodeficienze secondarie possono trarre beneficio dalle risposte mediate dalle cellule T. Al contrario, i vaccini con virus attivi non sono raccomandati nelle immunodeficienze secondarie. In merito alle vaccinazioni contro batteri capsulati, quelle per S. pneumoniae e H. influenzae sono raccomandate nei pazienti affetti da LLC.
2. Quali sono le terapie disponibili per le immunodeficienze secondarie e quali obiettivi si pongono?
La terapia sostitutiva con immunoglobuline (IgRT) ha radicalmente cambiato la storia naturale delle immunodeficienze secondarie e ha l’obiettivo principale di fornire protezione dalle infezioni tramite la somministrazione di anticorpi ad ampio spettro, prevenendo così le complicanze a lungo termine e migliorando la qualità della vita dei pazienti. Inoltre l’IgRT ha un’azione immunomodulante ad alte dosi nelle citopenie autoimmuni (Patel V, 2020).
Diversi studi hanno infatti dimostrato l'efficacia delle Ig nel ridurre l’incidenza di ricorrenze infettive e nell’ottenere un tempo libero da infezioni batteriche gravi più lungo (Patel SY, 2019).
I preparati con immunoglobuline umane sono ottenuti dal plasma dei donatori e sono costituiti da diversi anticorpi diretti contro svariati antigeni. Gli anticorpi sono rappresentati per la maggior parte da IgG ed in piccolissima quantità da IgM e IgA.
I dati clinici, laboratoristici e strumentali guidano la scelta di “quando” e “in chi” avviare la terapia sostitutiva con immunoglobuline, poiché debbono essere considerati il rischio di future infezioni, morbilità, mortalità e valutato il rapporto tra rischi e benefici (Jolles S,2017).
La terapia sostitutiva con Ig, per via endovenosa o sottocutanea, è raccomandata in caso di livelli sierici di IgG ≤400 mg/dl e infezioni gravi (una infezione maggiore o infezioni ricorrenti) e fallimento della profilassi antibiotica. La dose di immunoglobuline da infondere è di 400-600 mg/kg ogni 3-4 settimane e la sua variabilità dipende dai livelli sierici di IgG del paziente, dal numero di infezioni ricorrenti e dall’eventuale perdita renale o intestinale.
Le Ig possono essere somministrate per via endovenosa (IVIg), per via sottocutanea convenzionale (SCIg) o facilitata (fSCIg).
La somministrazione endovenosa permette di raggiungere rapidamente un alto picco di concentrazione plasmatica di Ig, utile per ottenere una rapida protezione dalle infezioni. Tuttavia, presenta alcuni limiti: richiede un accesso venoso, che in alcuni pazienti può rappresentare una difficoltà, ed avviene in ambiente ospedaliero per alcune ore. Ciò comporta un dispendio mensile di tempo, che equivale alla perdita di ore di scuola per i bambini o di lavoro per i genitori o i pazienti adulti. Le reazioni all’infusione possono essere variabili, solitamente da lievi a moderate (mialgia, febbre, brividi, cefalea, nausea, vomito, fenomeni trombotici), raramente gravi fino allo shock anafilattico.
La somministrazione sottocutanea convenzionale è una valida alternativa, perché praticabile anche a domicilio. Si utilizzano piccole pompe portatili che infondono le immunoglobuline in circa 1-2 ore e consentono di iniettare direttamente il farmaco a livello dell’addome, della coscia o del gluteo. La sede di infusione è il tessuto sottocutaneo, perciò la quantità di immunoglobuline introdotte è minore e l’assorbimento più lento, ma i livelli sierici si mantengono stabili più a lungo nel tempo. Nella maggior parte dei casi le reazioni sono locali (tumefazione e rossore nella sede di infusione) e col passare del tempo diventano meno frequenti. Ulteriore novità è rappresentata dalla somministrazione sottocutanea facilitata (fSCIg) che permette di allungare l’intervallo a 3-4 settimane grazie alla infusione di Ig preceduta da quella dell’enzima ialuronidasi che, creando una sorta di tasca nel tessuto sottocutaneo, permette di infondere volumi maggiori di farmaco (Danieli MG, 2016).
Le Ig sottocute hanno dimostrato un'efficacia paragonabile alle IVIg, con allo stesso tempo diversi vantaggi: livelli sierici di IgG più stabili, migliore qualità della vita ed efficienza in termini di tempo e costi sia per i pazienti che per il sistema sanitario, consentendo una maggiore autonomia al paziente e alla sua famiglia.
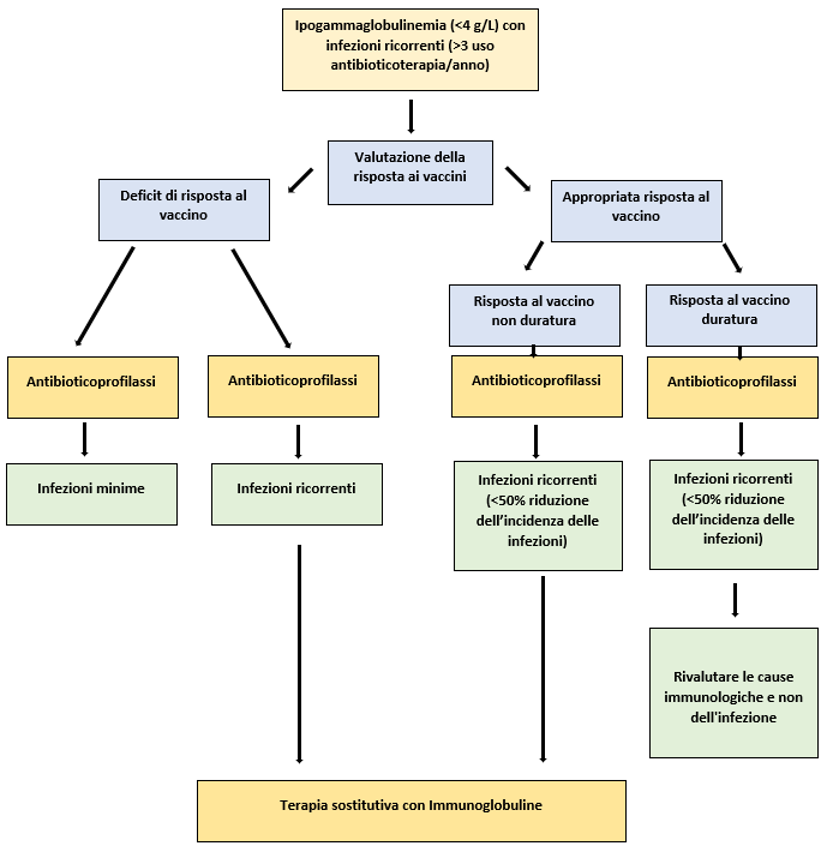
Figura 1- Protocollo iter diagnostico e terapeutico delle immunodeficienze secondarie
Nel nostro paziente è stata intrapresa alla diagnosi la terapia sostitutiva con SCIg (Hizentra® 40 ml/settimana), associata a profilassi antibiotica con azitromicina nei mesi invernali, con netta riduzione degli eventi infettivi. Da pochi mesi è stata modificata la terapia passando alla somministrazione con fSCIg (Hyqvia® 200 ml/3 settimane) per la ridotta disponibilità di alcuni preparati a base di Ig in seguito alla carenza di donazioni di plasma determinata dalla pandemia da COVID-19.
Bibliografia essenziale
- Patel SY, Carbone J, Jolles S. The Expanding Field of Secondary Antibody Deficiency: Causes, Diagnosis, and Management. Front Immunol 2019;10:33.
- Cinetto F, Neri R, Vianello F, et al. Subcutaneous immunoglobulins replacement therapy in secondary antibody deficiencie
- Real life evidence as compared to primary antibody deficiencies. PLoS One 2021;16(3):e0247717.
- Dhalla F, Misbah SA. Secondary antibody deficiencies. Curr Opin Allergy Clin Immunol 2015;15(6):50513.
- Patel V, Cowan J. Discontinuation of immunoglobulin replacement therapy in patients with secondary antibody deficiency. Expert Rev Clin Immunol 2020;16(7):7116.
- Jolles S, Chapel H, Litzman J. When to initiate immunoglobulin replacement therapy (IGRT) in antibody deficiency: a practical approach. Clin Exp Immunol 2017;188(3):33341.
- Danieli MG, Pulvirenti F, Rocchi V, et al. Selfadministered hyaluronidase-facilitated subcutaneous immunoglobulin therapy in complicated primary antibody deficiencies. Immunotherapy 2016;8(9):995-1002.