Stefano Gasparini, Martina Bonifazi, Lina Zuccatosta, Giacomo Spurio Vennarucci
Scenario clinico. Giungeva alla nostra attenzione un giovane di 25 anni, per l’approfondimento diagnostico di una dispnea ingravescente di recente comparsa. Dall’anamnesi emergevano un’abitudine tabagica attiva, oltre all’uso di sostanze voluttuarie - cannabinoli ed occasionalmente oppioidi endovenosi - per cui risultava in cura presso il SERT territoriale di riferimento. Il suo impiego di progettista nel settore dell’arredamento non lo esponeva a nocivi professionali od ambientali, e non erano note né familiarità per patologie polmonari né allergie. L’anamnesi patologica remota era caratterizzata dalla diagnosi, avvenuta nel 2012, di Morbo di Chron, trattato fino al 2019 con terapia biologica e successivamente con terapia steroidea sistemica, ed una spondilodiscite dorsale da Candida, in terapia con voriconazolo.
La storia clinica recente del paziente esordiva circa due mesi prima, con un accesso al Pronto Soccorso di un altro Nosocomio per insorgenza acuta di dispnea. Gli esami ematochimici all’ingresso erano caratterizzati da leucocitosi (17.903 globuli bianchi/mm3) ed un significativo incremento della proteina C reattiva (20,08 mg/dl), mentre l’emogasanalisi arteriosa mostrava insufficienza respiratoria parziale (PaO2: 55 mmHg, PaCO2: 36 mmHg, pH: 7,45); per tale motivo veniva trattenuto a ricovero.
Veniva eseguito un approfondimento con TC del torace ad alta risoluzione e con mezzo di contrasto iodato, che permetteva di escludere la presenza di embolia polmonare in atto, e che mostrava un quadro polmonare caratterizzato da noduli centrolobulari diffusi, piccole aree di oligoemia a mosaico ed intrappolamento aereo, in particolare ai lobi medio ed inferiori, ed aspetti di consolidamento con risparmio lobulare al lobo inferiore di destra. (Fig.1)
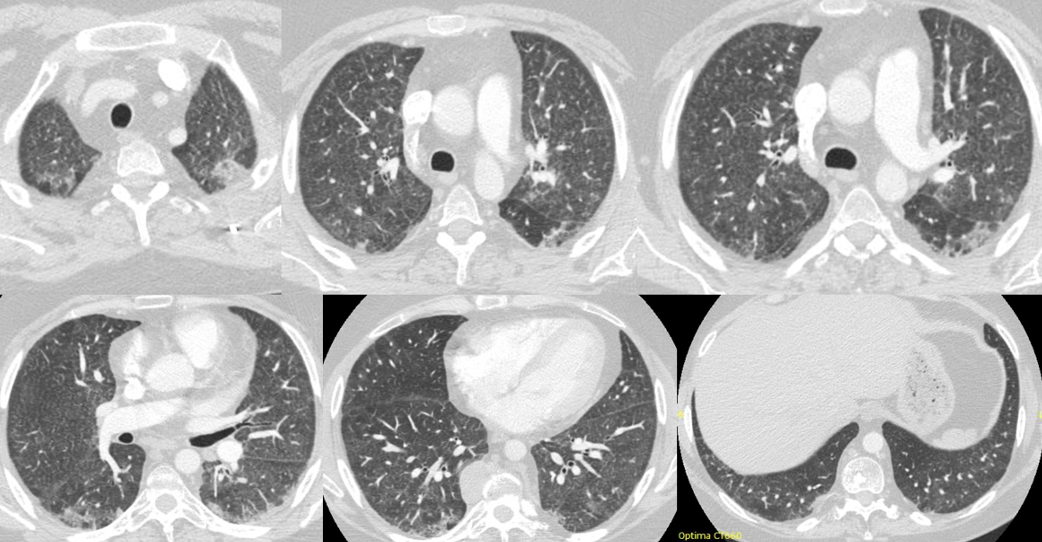
Fig. 1. Quadro TC polmonare caratterizzato da noduli centrolobulari, aree di oligoemia a mosaico, intrappolamento aereo e consolidazioni con risparmio lobulare
Il paziente veniva quindi sottoposto, in consulenza presso il nostro Servizio di Pneumologia Interventistica, ad una broncoscopia con lavaggio broncoalveolare, risultato negativo per la ricerca di germi comuni, mentre i terreni di coltura per bacilli alcool-acido resistenti avrebbero impiegato circa 40 giorni per dare il risultato definitivo.
I Colleghi decisero quindi di trattare il paziente con terapia antibiotica empirica ed incremento della terapia steroidea in atto, ottenendo un miglioramento clinico ed emogasanalitico tale da permettere la dimissione al domicilio.
Dopo due mesi, e circa dieci giorni dopo l’assunzione endovenosa di eroina, il paziente eseguiva un secondo accesso al Pronto Soccorso del medesimo Ospedale per nuova insorgenza di dispnea.
Alla luce del quadro emogasanalitico (PaO2: 78 mmHg, PaCO2: 32 mmHg, pH: 7,49 in FiO2 50%, P/F: 156), e della TC del torace, che mostrava un notevole incremento della componente a vetro smerigliato rispetto al precedente (Fig. 2), il paziente veniva trasferito c/o la nostra SOD di Pneumologia per proseguire l’iter diagnostico e terapeutico.
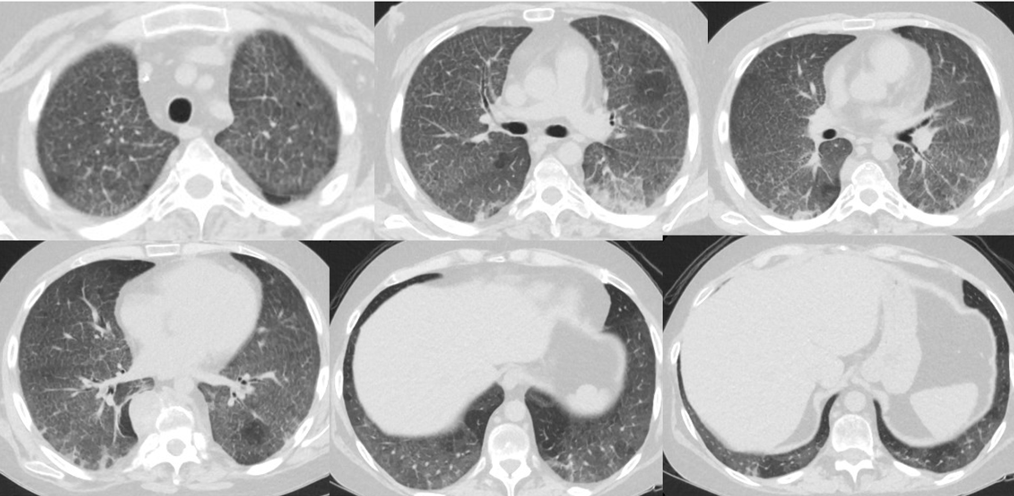
Fig.2. Quadro TC polmonare caratterizzato, rispetto al precedente, da incremento delle aree a vetro smerigliato
Il medico di reparto eseguiva un esame clinico completo, rilevando in particolare una frequenza respiratoria di 28 atti al minuto con utilizzo dei muscoli accessori della respirazione e la presenza all’auscultazione di crepitii diffusi in tutti i campi polmonari. Erano presenti gibbo e strie rubre a livello addominale, mentre all’esame obiettivo cardiologico non riscontrava alterazioni del ritmo, con battiti validi e pause apparentemente libere.
Alla luce di quanto osservato definiva il seguente problema clinico: severa insufficienza respiratoria parziale acuta in giovane paziente, fumatore attivo e prono ad abuso di sostanze stupefacenti, con anamnesi patologica di Morbo di Chron e spondilodiscite da Candida.
Il primo quesito diagnostico posto dal medico è stato il seguente: quale intensività di cura necessita il paziente in questa fase di patologia?
Per rispondere a questo quesito veniva eseguito un nuovo esame emogasanalitico per ridefinire il grado di compromissione degli scambi gassosi alveolari, con rilievo di una severa insufficienza respiratoria (P/F: 116). Le condizioni cliniche generali, lo stato di fatica respiratoria e la compromissione degli scambi gassosi alveolari, solo parzialmente corretti dall’inspirazione di una concentrazione di ossigeno del 50%, ponevano indicazione ad impostare un supporto ventilatorio meccanico. Le preservate condizioni neurologiche del paziente, che appariva vigile, orientato e collaborante, permettevano di ventilarlo in maniera non invasiva, mediante maschera facciale.
Veniva inoltre impostata una terapia di supporto, veniva incrementata la terapia steroidea in atto, e ovviamente interrotte le abitudini voluttuarie, seguendo le indicazioni terapeutiche dei Colleghi del SERT ospedaliero.
Nel frattempo, sei era reso disponibile il risultato degli esami colturali precedentemente eseguiti sul lavaggio broncoalveolare, con isolamento di Mycobacterium Xenopi, per il quale veniva impostata la terapia antibiotica specifica (claritromicina 500 mg bid, rifampicina 600 mg/die, e etambutolo 15 mg/kg/ml).
Dopo circa 15 giorni abbiamo potuto osservare un discreto miglioramento clinico ed emogasanalitico (PaO2: 71 mmHg, PaCO2: 35 mmHg, pH: 7,40 in FiO2 28%, P/F: 253).
Il paziente veniva sottoposto ad una nuova TC del torace, che mostrava un quadro di crazy paving, caratterizzato dall’ispessimento liscio a chiazze dei setti interlobulari con aree di opacamento a vetro smerigliato. La cessazione dell’abitudine tabagica e la terapia attuata avevano portato alla risoluzione della componente bronchiolitica, ‘smascherando’ il quadro TC sottostante (Fig. 3).
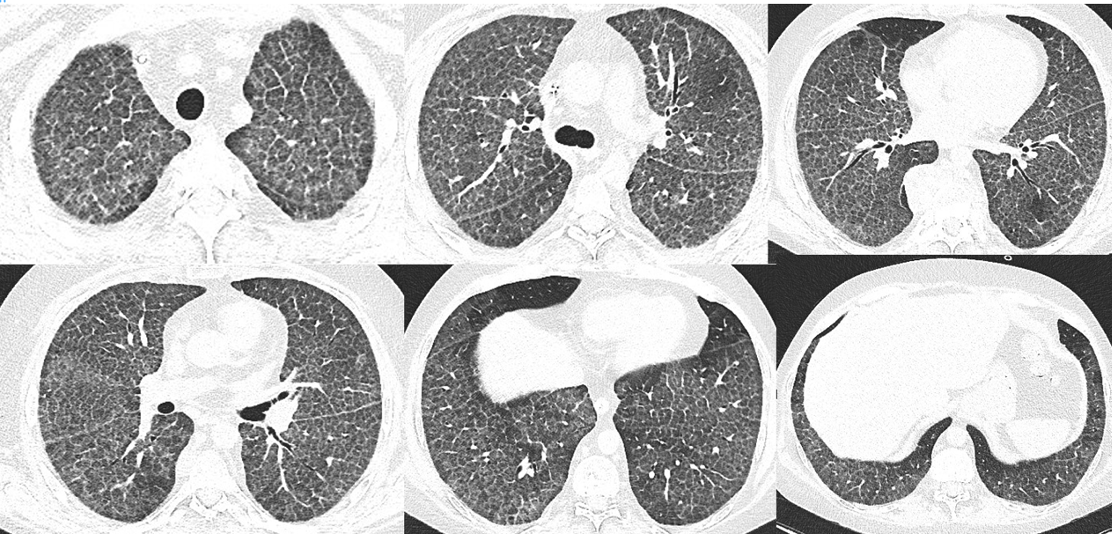
Fig. 3. Quadro TC polmonare di ispessimento liscio a chiazze dei setti interlobari ed aree di opacamento a vetro smerigliato
Sulla base di quanto raccolto era possibile formulare le seguenti ipotesi diagnostiche:
-Proteinosi alveolare polmonare: una condizione caratterizzata da un patologico accumulo alveolare di materiale proteinaceo PAS positivo, modesta infiammazione interstiziale e conseguente alterazioni degli scambi gassosi. Possibili trigger: immunodepressione cronica iatrogena, inalazione di sostanze esotossiche o infezione da micobattere
-Infezione polmonare: la terapia steroidea cronica potrebbe aver determinato una condizione di immunodeficienza iatrogena, con conseguente maggior suscettibilità alle infezioni polmonari. Sebbene il quadro TC di crazy paving risulti caratteristico di infezioni da Pneumocistis Jiroveci, non è possibile escludere a priori un quadro radiologico atipico da infezione da Micobattere.
-Sindrome emorragica polmonare: spettro sindromico caratterizzato da un sanguinamento parenchimale diffuso e bilaterale, idiopatico o secondario a vasculiti (Granulomatosi con poliangioite, granulomatosi eosinofila con poliangioite, Sindrome di Goodpasture), connetiviti (Lupus Eritematoso Sistemico, Artrite Reumatoide, Sclerosi Sistemica) o da farmaci.
-Manifestazione polmonare di Malattia Infiammatoria Cronica Intestinale: circa il 40% dei pazienti con MICI possono manifestare interessamento polmonare, solitamente caratterizzato da ispessimento delle pareti bronchiali ed intrappolamento aereo.
In base a queste considerazioni il medico ha richiesto le seguenti indagini di approfondimento:
-Elettroforesi proteica, Fattore Reumatoide, anticorpi anticitrullina, Anca, ANA, ENA, anticorpi anti-Smith, anti-Scl70 ed anti-centromero, per escludere la presenza di patologie autoimmuni: risultate negative.
-Valutazione ecocardigradica: cinetica globale cardiaca conservata, frazione di eiezione 60%, PAPs di 30+5 mmHg.
-Prove di funzionalità respirtoria: deficit ventilatorio restrittivo di grado moderatamente elevato, con severa riduzione della DLCO.
Il quesito diagnostico posto dal medico è il successivo: è necessario proseguire con indagini cito-istologiche per raggiungere il corretto inquadramento diagnostico?
Gli esami condotti hanno permesso di escludere con ragionevolezza la presenza di una eziologia autoimmune alla base delle manifestazioni cliniche del paziente. Il lavaggio broncoalveolare eseguito non mostrava isolamento di P. Jiroveci. L’anamnesi farmacologica era muta per terapia anticoagulante ed antiaggregante.
Per poter verificare il sospetto di proteinosi alveolare polmonare, o come ipotesi alternative un danno parenchimale diffuso da inalanti ed infine una localizzazione polmonare di MICI, si decideva per eseguire una nuova broncoscopica, con esecuzione di un nuovo lavaggio broncoalveolare e biopsie polmonari transbronchiali mediante criosonda in corrispondenza del lobo inferiore di destra.
La criobiopsia transbronchiale è una procedura broncoscopica impiegata nel work-up diagnostico delle patologie infiltrative diffuse. Si configura come metodica alternativa alla biopsia chirurgica, essendo caratterizzata da un profilo rischio-beneficio migliore, e pertanto è attualmente proposta come prima indagine, riservando l’approccio chirurgico in caso di inconclusività dei risultati.
Il liquido di lavaggio broncoalveolare aveva un aspetto macroscopico lattescente, permettendo di escludere l’ipotesi di un danno alveolare diffuso e rappresentando un importante elemento a favore della diagnosi di proteinosi alveolare polmonare (Fig. 4).
L’esame istologico mostrava la presenza di abbondante materiale eosinofilo PAS positivo all’interno degli alveoli, con disegno alveolare conservato. Lo spazio era occupato da materiale eosinofilo omogeneo, scarsamente cellulato. Anche l’interstizio appariva sostanzialmente conservato, con solo scarso infiltrato linfocitario. La ricerca per agenti eziologici infettivi era negativa (Fig.5)
Era presente inoltre materiale estraneo interstiziale-perivascolare di aspetto cristallino, determinante una reazione granulomatosa, con vasi limitrofi caratterizzati da alterazioni della parete e del decorso (Fig.6).
Quanto osservato appariva compatibile con un quadro di proteinosi alveolare polmonare e presenza di fenomeni di trombosi e rivascolarizzazione dovuti a uso endovenoso di sostanze esogene con evocazione di una reazione granulomatosa (Box proteinosi alveolare polmonare).

Fig. 4 liquido di recupero del lavaggio broncoalveolare
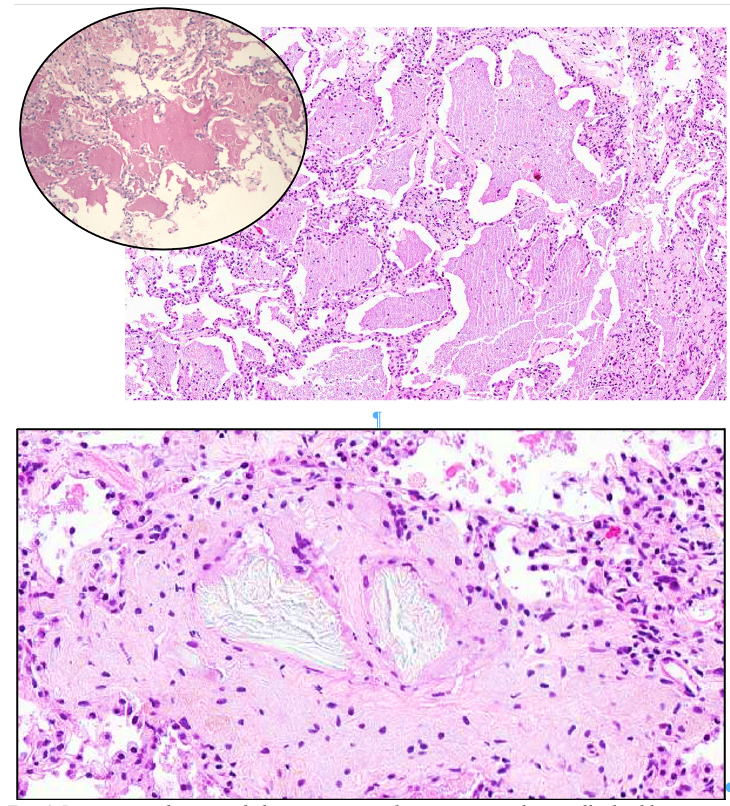
Fig. 5. Presenza di materiale proteinaceo all’interno degli spazi alveolari. Nel dettaglio la colorazione PAS positiva
Fig. 6. Lesioni granulomatose di dimensioni grossolane con materiale cristalloide al loro interno
La proteinosi alveolare polmonare (PAP) è una sindrome polmonare rara, caratterizzata dal patologico accumulo di surfactante e lipidi a livello degli alveoli polmonari, determinante l’alterazione degli scambi gassosi.
Decritta per la prima volta nel 1958, ha una prevalenza di 3,7-6,2 casi per milione di abitanti, con interessamento doppio degli uomini rispetto alle donne. Si osservano due picchi di incidenza di malattia: un primo fra i 25 e 45 anni ed uno dopo i 75 anni di età.
La PAP viene classificata in base al meccanismo d’azione in:
1) Primaria: causata da un’alterazione del signaling del GM-CSF. Questo determina un’alterazione della maturazione dei macrofagi polmonari, che non sono in grado di degradare correttamente i lipidi del surfactante, con conseguente accumulo alveolare.
Il 90-95% delle PAP sono primarie, e sono legate alla presenza di anticorpi classe IgG rivolti verso diversi epitopi del recettore di GM-CSF, con concentrazioni > 5 microgrammi/dl.
2) Secondaria (5-10% dei casi di PAP): dovuta ad alterazione qualitativa o quantitativa dei macrofagi alveolari, legate a sindromi mielodisplastiche, infezioni croniche, inalazione di sostanze tossiche, o condizioni di immundeficienza.
3) Congenita ed Ereditaria (< 1%): osservata principalmente in età pediatrica, è legata a mutazioni che determinano alterazioni nella produzione del surfactante, nel trasporto intracellulare dei lipidi coinvolti nella produzione dello stesso o mutazioni che determinano alterazioni nello sviluppo polmonare. Questa forma rappresenta circa il 2% delle proteinosi alveolari polmonari.
La forma ereditaria è dovuta alla mutazione dei geni CSF2RA e CSFR2RB, coinvolti nella formazione delle subunità del recettore di GM-CSF
4) Idiopatica (1%): qualora non siano presenti anticorpi rivolti contro GM-CSF e non sia possibile inquadrare possibili cause di secondarietà (Tab. 1)
|
Tipi di PAP e cause |
|
Primaria (o auto-immune) (90-95% dei casi) Dovuta ad alterazione del signalling della via del GM-CSF da anticorpi rivolti contro il recettore GM-CSF |
|
Secondaria (5-10% dei casi) Causata da: -disordini emopoietici (sindromi mielodisplastiche [deficit GATA2], deficit plasmacellule, leucemia mieloide, linfoma -alterazioni immunologiche (immunodeficienza severa combinata [SCID], ipogabbaglobulinemia, anemia di Fanconi, sd. Di Bechet, trapianto di midollo osseo, trapianto polmonare -infezioni: Nocardia, Pneumocistis Jiroveci, CMV, HIV -inalanti (polvere di silicio, alluminio, titanio, fibre vegetali, derivati del benzene) -farmaci: micofenolato mofetile, leflunomide, imatinib, busulfano, ciclosporina, fentanyl, sirolimus, silicone di impianti |
|
Congenita ed Ereditaria (< 1% dei casi) Dovuta a mutazione recessiva a carico del signalling GM-CSF (CSF2RA, CSF2RB), mutazioni a carico dei geni del surfactante (SFTPB, SFTPC, ABCA3, NKX-1 [TTF-1]) |
|
Idiopatica (<1%) In assenza di anticorpi anti- GM-CSF, di bacjground secondario o mutazioni note |
Tab. 1. [Modificata] Classificazione della proteinosi alveolare polmonare. ‘Pulmonary alveolar proteinosis in adults: pathophysiology and clinical approach’ Kumar et al. Lancet Respir Med 2018; 6: 554–65
Quesito prognostico: qual è l’evoluzione della proteinosi alveolare polmonare?
L’evoluzione della patologia è imprevedibile. Sono descritti in letteratura casi di risoluzione spontanea (17% dei casi), avvenuti dopo la cessazione dell’esposizione a verosimili fattori favorenti (fumo di sigaretta e polveri). La proteinosi alveolare si manifesta con un range di gravità variabile, fino a configurare un quadro di severa insufficienza respiratoria – come nel caso del nostro paziente – che può condurre all’exitus. La sopravvivenza attuale a 5 anni è del 95%. Non esistono attualmente predittori di gravità della patologia.
La principale complicanza è l’insorgenza di infezioni secondarie, responsabili di circa il 20% delle morti dei pazienti con PAP. I patogeni più frequentemente coinvolti sono gli Streptococchi, Haemophilus spp. e le Enterobacteriaceae, ma possono essere presenti patogeni come micobatterio tubercolare ed atipici, Nocardia spp. e Aspergillus spp. Bisogna sospettare la presenza di una sovrinfezione in caso di comparsa di sintomatologia atipica, come toracalgie ed emottisi.
Un’altra possibile complicanza, legata alla cronica stimolazione infiammatoria a carico del parenchima polmonare, è l’insorgenza di fibrosi polmonare, con pattern ad alveare.
Il medico di reparto ha così completato l’inquadramento diagnostico, ponendo la diagnosi di Proteinosi Alveolare Polmonare secondaria ad infezione da Mycobacterium Xenopi.
Si è infine posto il seguente quesito terapeutico: quali sono attualmente le opzioni terapeutiche disponibili per la proteinosi alveolare?
Nel caso delle forme secondarie il primo intento terapeutico deve essere la gestione e possibilmente la rimozione della causa scantenante. In tal senso il paziente proseguirà la terapia antibiotica mirata in atto per un periodo minimo di 12 mesi. Alla terapia eziologica può essere eventualmente associato il lavaggio polmonare profondo, che risulta invece l’attuale gold standard nelle forme primarie: si trattata di una procedura eseguita in anestesia generale, caratterizzata dall’intubazione selettiva monolaterale e instillazione di 20 fino a 40 litri di soluzione fisiologica sterile, che vengono progressivamente recuperati, ottenendo la progressiva clearence del materiale proteinaceo alveolare. La procedura viene quindi ripetuta dopo almeno 14 giorni al polmone controlaterale.
Non esistono attualmente prove univoche sull’efficacia della terapia immunomodulante (rituximab), mentre sono in studio, per i pazienti con forme autoimmuni, terapia inalatorie con GM-CSF ricombinante, non ancora disponibili per la pratica clinica.
Raccomandazioni finali
- La proteinosi alveolare polmonare (PAP) è una sindrome caratterizzata dall’accumulo di surfactante negli alveoli e nelle vie aeree terminali che può determinare insufficienza respiratoria ipossiemica
- Il surfactante è sintetizzato e secreto dalle cellule epiteliali tipo II alveoli, ed il suo catabolismo è dovuto ai macrofagi alveolari
- La PAP autoimmune è la forma più frequente (90-95% dei casi) ed è dovuta ad IgG rivolte contro i recettori di GM-CSF, con alterazione della via di maturazione dei macrofagi alveolari, che risultano disfunzionali.
- L’aspetto macroscopico lattescente del liquido di lavaggio broncoalveolare (BAL) è suggestivo di Proteinosi Alveolare Polmonare.
- Il lavaggio polmonare profondo è attualmente il gold standard terapeutico, sebbene siano in corso trial clinici promettenti per il trattamento della forma autoimmune
Letture consigliate
- Rossi SE et al., Crazy Paving Pattern at Thin-Section CT of the Lungs: Radiologic-Pathologic Overview. RadioGraphics 2003; 23:1509-1519
- Jouneau S et al., Pulmonary Alveolar Proteinosis. Respirology (2020)
- Salvaterra E, Campo I. Pulmonary Alveolar Proteinosis: from Classification to Therapy. Breathe, June 2020 Vol 16 No 2
- Kumar et al. Pulmonary Alveolar Proteinosis in Adults: Pathophysiology and Clinical Approach. Lancet Respir Med 2018; 6: 554–65
- Santos FG et al. Alveolar Proteinosis due to Toxic Inhalation at Workplace. Respiratory Medicine Case Reports 31 (2020).